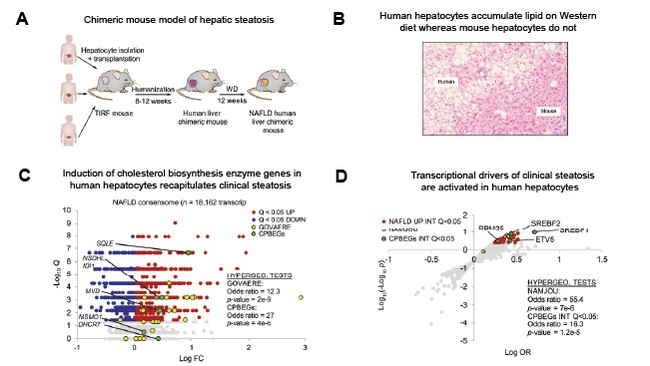
Although mouse models are widely used to study human disease, many fail to recapitulate the pathophysiology of clinical disease. Validation against clinical datasets is therefore a critical component in the development of such models. Our laboratory collaborators developed a chimeric mouse model of human disease (Image, A) that recapitulated histological features of human hepatic steatosis after 12 w on western diet, with steatosis being confined to the human hepatocytes (Image, B). To validate this model we used archived clinical steatosis datasets to generate a transcriptomic consensome, and showed that the transcriptional regulatory profile of the chimeric human hepatocytes strongly recapitulated that of clinical steatosis (Image, C&D).
Dr Neil McKenna leads the team. Trained as a cell biologist, he has 20 years experience in data sharing and re-use initiatives. Reflecting this experience, he led the first data repository in the US to mint digital object identifiers as unique identifiers for datasets. He currently leads the Signaling Pathways Project (SPP), a trans-omics knowledgebase for cellular signaling pathways. His career goal is to give datasets parity of esteem with research articles.
Dr Scott Ochsner joined Dr McKenna's group in 2009 after graduate research in molecular reproductive biology. He's never met a dataset he didn't want to put through R and is happy to do the leg work required to throw a lens on to an RNA-Seq dataset to make sense of its biology.
Period of time and goals
This work was carried out in 2019 - 2020.
Although non-alcoholic fatty liver disease (NAFLD) affects a quarter of the world's population, standard of care is based largely on lifestyle modification. There is an urgent need therefore for the development of reliable models of clinical NAFLD to test novel pharmacological interventions. The goal of this project was to use archived clinical transcriptomic datasets to validate a novel chimeric mouse model of NAFLD.
What data sharing and/or reuse practices has your team adopted?
Using our previously-described consensome algorithm (Ochsner et al (2019) Sci Data 6, 252) we generated a clinical NAFLD consensome, which ranked ~18,100 human genes according to the frequency with which they were upregulated or downregulated across 22 independent, archived clinical NAFLD case-control transcriptomic datasets.
What data sharing or reuse practices would you recommend all researchers adopt, and why?
With respect to sharing of clinical datasets, it is particularly important to provide appropriately de-identified clinical metadata. For example, we would have ideally liked to use the archived NAFLD datasets to identify consensus gene expression patterns that predicted severity of NAFLD. Unfortunately none of the datasets included information on disease severity. With a modest amount of additional effort, inclusion of indices of clinical severity would have made our analysis even more compelling.
What do you think is compelling about how you shared or reused data?
Remarkably, our collaborators had shown that in response to western diet, the chimeric human hepatocytes accumulated lipid, whereas the mouse hepatocytes did not (Image, B). Scrutinizing RNA-Seq data from each cell type, we found that cholesterol biosynthesis pathway enzyme genes (CPBEGs) were expressed at higher levels in the human hepatocytes than the mouse hepatocytes. The question was, how closely did this reflect the situation in clinical NAFLD? Validating the model, we showed that CBPEGs were also robustly enriched among the highest confidence clinical NAFLD consensome-induced genes. CTs. Taken together, our analysis demonstrated a robust convergence between the elevated expression of CBPEGs in clinical NAFLD and in the steatotic human hepatocytes in the chimeric mice.
Meta-analysis is widely used to identify commonalities across existing 'omics studies. However, in order to gain credence and acceptance by reviewers and the broader research community in turn, a meta-analysis must show that it accurately recapitulates the results of independent related studies.
With respect to the NAFLD consensome, we were therefore required to demonstrate to reviewers that it assigned elevated rankings to genes whose expression levels had been shown in independent studies to contribute to NAFLD pathogenesis. To do this we identified a recently published study by Govaere and colleagues (Sci. Transl. Med. (2020) 12, eaba4448) that generated transcriptomic dataset from a NAFLD clinical cohort and that was not included in our study. This study resolved a 25-gene signature that predicted clinical NAFLD severity. Going to the reliability of the NAFLD consensome, hypergeometric analysis showed that this signature was robustly enriched among the highest confidence NAFLD consensome-induced genes.
There are two critical components to replicating our approach. The consensome code has been deposited in github and is freely available to all users. Equally critical is the curation and quality control of the underlying datasets. To this end we have published a resource article (JAMIA (2016) 24, 388-393) that describes in detail how datasets are retrieved from archives, processed to extract data points, then subjected to metadata enrichment and gap filling.
Despite their transformative impact on discovery-driven research, modest rates of public archiving, and sporadic compliance with metadata annotation standards, have restricted the full potential of transcriptomic datasets in the field of eukaryotic cellular signaling. The net result has been to promote in this field a research information cycle from which transcriptomic datasets have been largely excluded as independently citable resources. Given the platform we would tell researchers that there is a vast universe of archived omics datasets that collectively illuminate relationships between signaling pathways and their target genes in human disease that have not yet been explored in the research literature. By making greater use of these datasets we hope that researchers can help bring data re-use more into the mainstream so that archived, re-used datasets can gain the recognition they deserve as essential components of the research life cycle.
